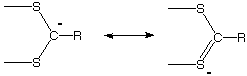Chemical reaction mechanisms are generally "proven" by the Sherlock Holmes principle:
"It is an old maxim of mine that when you have excluded the impossible, whatever remains, however improbable, must be the truth."(from "The Adventure of the Beryl Coronet." Similar statements occur in many other Holmes stories.)
So actually one designs experiments or arguments to disprove alternative mechanisms rather than to prove a proposed mechanism.
Of course logically this requires testing every possible mechanism, which is clearly impossible. But some mechanisms are so energetically demanding as to be ludicrous (e.g. dissociating into a set isolated atoms and reassembling in the pattern of the products), and generally one is reluctant to consider complicated schemes when a simple one will do (this is the somewhat naive logical principle called Ockham's Razor).
[Actually, to be brutally frank, rigorous logic does not often play the dominant role in developing theories. Scientists commonly just blunder along with half-baked ideas until they are forced by some kind of accident to alter them. Still, when we come to deciding what theories we should believe, it is admirable to apply logic.]
For substitution reactions at tetravalent carbon one can imagine a Concerted process, in which the new bond is formed as the old one is broken, and two classes of interactions with intermediates: Dissociation/Association with a trivalent carbon intermediate, and Association/Dissociation with a pentavalent carbon intermediate. For the reactions with intermediates either the first or the second step (formation or destruction of the intermediate) might be rate-limiting. Thus in a particular case one could imagine 5 relatively simple mechanism, and hope that eliminating all but one will leave the true mechanism (i.e. that the true mechanism is not a more complex one).
In fact really careful study over more than half a century has shown that there are indeed complicating subtleties, involving for example what the other ion paired with EtO- might be doing, but we will stick with the cruder, mostly correct generalizations.
The simple observation that the rate of EtO- + EtBr depends on the concentration of the EtO- nucleophile, excludes D/A with the first step rate-limiting, because in this mechanism the nucleophile has not yet come on the scene when the rate-determining transition state forms.
D/A with the second step rate-limiting is implausible, since this would require the trivalent Et+ cation intermediate to react with Br- (and return to starting material) more easily than it reacts with EtO- to give product. The pKa of HBr (-4.7) is smaller than that of HOEt (15.9) by >20 pK units, i.e. EtO- has a >1020 times larger equilibrium constant for reacting with H+ than does Br-. Even though Et+ is not H+, and differences in activation energy are not the same as overall energy differences (but see Hammond Postulate), it would be truly astonishing if Br- overcame this 1020 fold handicap to react more rapidly than EtO- with Et+.
Also because of the greater stability of Br-, A/D with the second step rate-limiting would be unlikely, since a pentavalent anionic intermediate should not lose EtO- in preference to Br-.
Kinetic order and reasonable logic have excluded three of the simple mechanisms leaving only the concerted mechanism, in which the pentavalent carbon species is a transition state (saddle point on the potential energy surface with an energy maximum along the reaction coordinate) and the A/D mechanism with the first step rate-limiting (and a local energy minimum along the reaction coordinate). Since a tiny energy dimple at the crest of the concerted path would constitute the intermediate of an A/D mechanism, these two candidates may not be trivial (or very important) to differentiate experimentally.
If pentavalent carbon anions are stable enough to be considered important intermediates, there should, among the very large number of organic compounds that have been prepared, be some examples of pentavalent carbon that are stable enough to observe directly. (Again this is an argument of plausibility, not of rigorous logical proof.)
Are there any examples of "hypervalent" carbon?
Yes, there are a few examples of pentavalent carbon compounds, but not such as you would be dying to write home about because they are so impressively stable. Seven years ago an example based on an X-ray structure determination was proposed by Akiba, Yamashita, Yamamoto, and Nagase in the Journal of the American Chemical Society (vol. 121, pp. 10644-5, 1999). As we show below this example is bogus.
They carried out the following reaction and crystallized a product with a carbon cation squeezed between adjacent ROCH3 nucleophiles ("nucleus lovers", groups with high HOMOs) within the same molecule:
[Note that an SN2 reaction is used to add CH3+ from Me3O+ (an hypervalent ion) to the carbonyl oxygen of the ester, and that the crazy B2F7- anion is formed from using the vacant orbital of a BF3 to stabilize an unshared pair of BF4-. The BF3 must have arisen from another BF4- losing an F- to some other LUMO.][Note also that there is no way the molecule could be planar, since the ester group would have to twist to avoid the methoxyl groups.]
Below is the structure of the crystallized product as determined by x-ray diffraction and shown in the JACS paper:
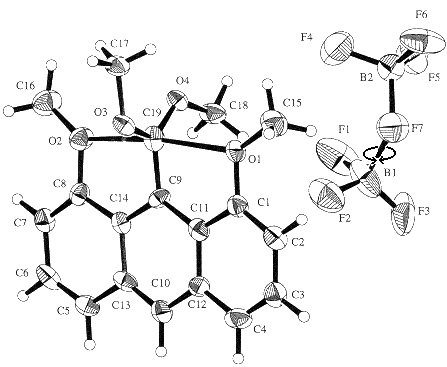
The flat "cationic" carbon (C19) is sandwiched between O1 and O2 of the two methoxyl groups and is nearly equidistant from them, 2.45 and 2.43 Ĺ, much further than normal C-O bonds(1.43 Ĺ), but much closer than van der Waals contact (3.25 Ĺ).
C19 is certainly drawn as pentavalent with lines denoting bonds. But maybe the C19-O1 and C19-O2 distances are short simply because of how the ring skeleton holds these atoms.
Is there really an attractive, bonding force between C19 and the adjacent oxygens?
The answer proposed in the JACS paper was "Yes" (but see below). The C19-O1 and C19-O2 distances (average 2.44Ĺ) are a little shorter than C9...C1 and C9...C8 (average 2.51Ĺ), meaning that the C1-O1 and C8-O2 bonds are slightly bent inward. It was proposed that this bending was caused by bonding attraction between the O atoms and C19.
Of course it's not too surprising that the unshared pairs on O1 and O2 might be stabilized by mixing with the LUMO on C19, but it might have been that the lowest energy form had C19 bent toward one of the two flanking O atoms, rather than being symmetrical, so as to be more tetravalent than pentavalent.
One should be cautious in interpreting this kind of X-ray data, since X-ray structures are not instantaneous pictures of individual molecules, but superposition of instantaneous structures of all molecules in the crystal over a long time. That is, if some molecules had C19 bent one way, and some bent the other, the superposition might look symmetrical when the individuals were not.But there is a clue that this is not happening here. The illustration is a style called an ORTEP plot (for Oak Ridge Thermal Ellipsoid Parameter), drawn with a program developed about 40 years ago at Oak Ridge National Laboratory by a former Yale chemist. Shapes of the atom ellipsoids, which in this case are drawn to include 30% of the total electron density, show how varied the positions of individual atoms are. Notice that atoms F1-F6 are elongated in a direction perpendicular to their bond to B, suggesting that there is a little bit of back and forth rotation (see arrow) of BF3 groups about the B-F7 bonds. Similarly the ellipsoids for C16 and C18 show the directions in which these methyl carbons vibrate. But the C19 ellipsoid is not elongated horizontally, showing that C19 is not moving back and forth between O1 and O2.
In a brief letter to Chemical & Engineering News (Feb 14, 2000, p. 13) Professor J. B. Levy of the University of North Carolina-Wilmington disputed the above interpretation of the x-ray results. He reported that his (unspecified) calculations show no special bonding interaction between the cationic carbon and the flanking methoxy oxygens and that even without a carbon cation the methoxy groups bend in toward the center of the molecule! Thus it would be a mistake to interpret the x-ray demonstration of bending as evidence for the supposed pentavalent bonding.
Though Levy doesn't say so, one might
suppose that in the observed eclipsed, planar conformation,
repulsion between the methyl group and the adjacent
(ortho) hydrogen atom would distort angles a, b, and
c to give a value that one might expect for a
normal ether. In the "pentavalent" compound above,
where X is a carbocation, a-b = 11° and c
= 122°. But in 19 examples of this grouping where X=H
(taken from a computer data base of crystal structure data)
a-b ranges from 7° to 15° (average
11°!), and c ranges from 116° to
119°. Thus the bending can easily be explained in terms
of steric hindrance of the methoxy methyl group. In fact a calculation (MacSpartan,
STO 6-31*) on phenol (hydroxybenzene) shows
a-b = 5°, c = 110.7°. Even without
the methyl group, eclipsing about the C-O bond causes the
same type of distortion.
and
c > ~110°,
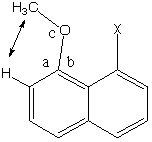
The compound shown above was tailored to optimize the chances for observing pentavalent carbon (very low LUMO on central carbon, reasonably high HOMOs on the flanking oxygens). That the value of a-b for this molecule are no larger than for cases without bonding between O and X makes it seem likely that any C19-O1 and C19-O2 bonding in the "pentavalent" carbon is so weak that it would never survive against the bias of entropy if the rest of the molecule did not keep the atoms from moving apart. It also seems likely that if the pentavalent molecule were an anion (as in EtO- + EtBr) rather than a cation, it would not be stable.
Because it doesn't make too much difference anyway, we will assume that the paucity of examples of stable pentavalent carbon means that normal SN2 reactions go by a concerted mechanism rather than by an A/D mechanism, but the possibility of even a few examples of stable pentavalent carbon would make one cautious about being doctrinaire.
Problem: It is a challenge to draw a "Kekulé" structure for the pentavalent species, and to decide where to place formal charges. This is more an opportunity to think about what such structures mean than a problem with a single well-defined answer.
Note on hypervalency. We have already encountered many cases where an atom is "hypervalent", that is, where it forms more bonds than one would expect given its normal valency. One type of case is where the atom uses an unshared pair of electrons to mix with someone else's low LUMO, e.g. (CH3)3O+ or NH4+. In such cases the atom assumes a formal positive charge. These are called "-onium" ions, here oxonium, and ammonium.Another example would be where an atom has vacant valence shell orbitals that it can use to accept electron pairs from atoms with high HOMOs. One can consider the stabilization of a carbon anion by an adjacent S atom with vacant d-orbitals in terms of a valence structure with trivalent S:
Pentavalent carbon is a little different since tetravalent carbon has neither unshared pairs nor vacant valence-shell orbitals.
[Return to text]
|
|
|